FDA Criticized as COVID Tests Still not Accurate, But U.S. Starts Second Lockdown Anyway
 By Brian Shilavy, Editor, Health Impact News
By Brian Shilavy, Editor, Health Impact News
The corporate media news cycle this week is once again promoting fear in the American population by claiming that COVID cases are again on the rise in “hot spots,” prompting calls for more lockdowns and other measures, including many states now requiring people to wear face masks in public.
Two key pieces of information are missing from almost all of these reports in the corporate media: death rates (even by their own statistics) are NOT increasing but holding steady or even decreasing, and inaccuracy with the tests themselves are still widespread.
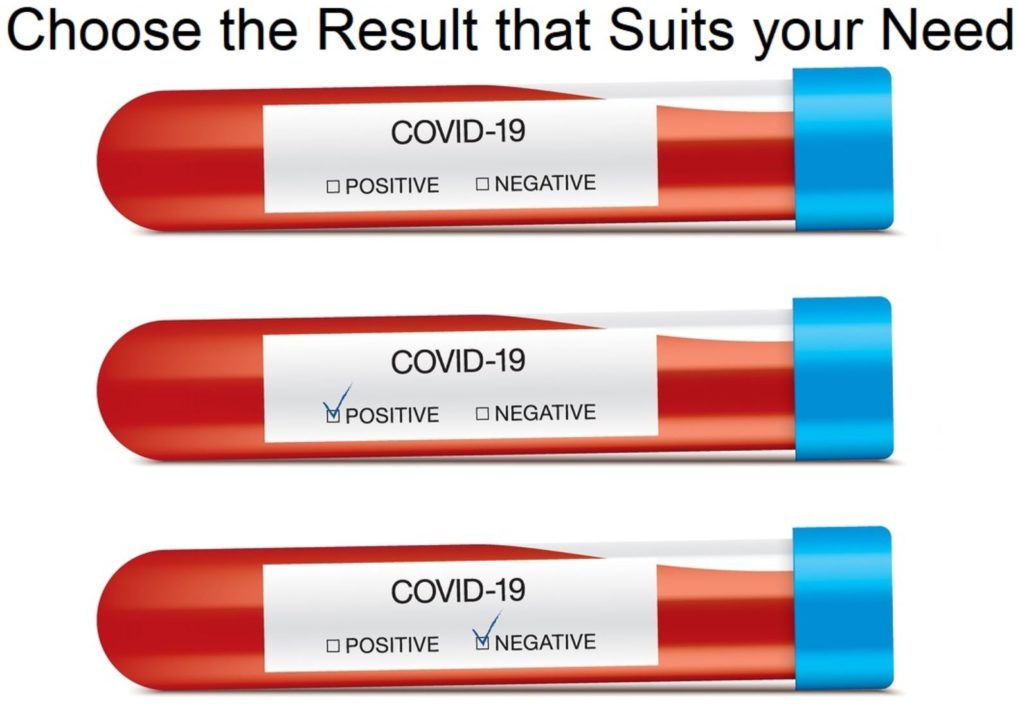
A report earlier this month out of Wichita Falls, Texas, for example, revealed that testing of residents and staff at a medical facility revealed many positive results, but since none of them were sick, they retested 20 of them, and the second test result was negative in all 20 of them.
Widget not in any sidebars
In a Facebook post Monday night Sheridan Medical Lodge reported that 78 residents and staff members tested positive for COVID-19 during routine testing conducted on May 25 however after retesting 20 staff and residents the test came back negative.
According to the post, 46 residents and 32 staff members tested positive during routine facility testing.
All positive cases were asymptomatic. According to the post, it is unusual for cases at a nursing facility to not show any symptoms which prompted a retest of 20 residents and staff.
The results of all 20 retest have come back negative for COVID-19. (Source.)
The national corporate media franchises would probably never publish something like this, because it doesn’t fit their narrative for the Plandemic.
They do report inaccuracies with testing, however, if it does fit their narrative, meaning that tests are inaccurate in the sense that there should be more positive results, especially if it is a test promoted by President Trump, such as Abbott’s fast COVID-19 test.
FierceBiotech, a pharmaceutical marketing publication, reported:
In mid-May, the Food and Drug Administration issued a rare public warning about an Abbott Laboratories COVID-19 test that for weeks had received high praise from the White House because of its speed: Test results could be wrong.
The agency at that point had received 15 “adverse event reports” about Abbott’s ID NOW rapid COVID-19 test suggesting that infected patients were wrongly told they did not have the coronavirus, which had led to the deaths of tens of thousands of Americans. The warning followed multiple academic studies showing higher “false negative” rates from the Abbott device, including one from New York University (NYU) researchers who found it missed close to half of the positive samples detected by a rival company’s test.
But then, in a move that confounded lab officials and other public health experts, a senior FDA official later that month said coronavirus tests provided outside lab settings would be considered useful in fighting the pandemic even if they miss 1 in 5 positive cases—a worrisome failure rate.
The FDA has now received a total of 106 reports of adverse events for the Abbott test, a staggering increase. The agency has not received a single adverse event report for any other point-of-care tests meant to diagnose COVID-19, an agency spokesperson said.
In a statement, Abbott Laboratories said the NYU research was “flawed” and “an outlier,” citing studies with higher accuracy rates.
Though the Abbott rapid test is one of over 100 COVID-19 diagnostic tests to receive FDA emergency use authorization during the pandemic, President Donald Trump has featured the product in the White House Rose Garden, and the Department of Health and Human Services’ (HHS’) preparedness and response division has issued more than $205 million worth of contracts to buy the test, according to federal contract records. (Source.)
About the only truth the public can ascertain from all of these conflicting reports is, the COVID tests simply are not accurate.
As the FierceBiotech publication noted, over 100 COVID-19 diagnostic tests have received FDA emergency use authorization to fast-track them and bring them to market.
That means ZERO COVID tests have gone through the full approval process to bring a test to market, which would normally take years.
As I reported back in May, someone contacted me who has 43 years of clinical diagnostics experience. In addition to being a Med Tech at one of the New York City metropolitan area’s largest reference laboratories, this person spent 25 years of that 43 working for medical device manufacturers as a biomedical field service engineer and technical consultant. They installed, repaired, troubleshot and validated laboratory instrumentation.
This person wishes to remain anonymous, but this is what they wrote:
FDA emergency Use Authorization and COVID-19 Testing
by a 43-year Veteran of Clinical Diagnostics
Since the beginning of the COVID-19 “Pandemic”, much of government and the media’s focus has been on the need for more testing.
The purpose of this brief article is to examine the FDA’s Emergency Use Authorization (EUA) and the effect these authorizations have on the reliability of test results in identifying positive/negative samples for COVID-19.
Per the FDA website:
Under section 564 of the Federal Food, Drug, and Cosmetic Act (FD&C Act), the FDA Commissioner may allow unapproved medical products or unapproved uses of approved medical products to be used in an emergency to diagnose, treat, or prevent serious or life-threatening diseases or conditions caused by CBRN (Chemical, Biological, Radiological and Nuclear) threat agents when there are no adequate, approved, and available alternatives. (Source.)
The key word in this statement is “unapproved.”
Medical device manufacturing is one of most highly regulated industries in the US. The FDA approval process for a medical device takes years.
To date there are no COVID-19 diagnostic tests being used that have completed a full FDA approval process.
The reason they have not received FDA approval is because the safety and effectiveness of the product has not been proven. Many of these tests have been developed at least since the SARS outbreak of 2000-2004.
Refer to the above FDA website video explaining the EUA process.

Pay particular attention to the statement:
It is not in the best interest of Americans for the FDA to allow the use of a test that doesn’t work as it should. False test results can contribute to the spread of an infectious disease like COVID-19.
In reviewing the list of In Vitro Diagnostics products on the FDA website that have received Emergency Use Authorizations, the FDA is contradicting its own claim and is authorizing the use of diagnostics tests that produce false results.
There is a very easy way to confirm this statement, if you know where to look.
From the list of In Vitro Diagnostic products that have received a EUA, select any manufacturer. Go to that manufacturer’s website. Select the test from the products listing.
Look for the “package insert”. The package insert explains everything you need to know about the test, including its intended use, performance and interpretation of results.
Using the ROCHE cobas® SARS-CoV-2 test from the list, go to the Roche website and read the emergency use statement:
Results are for the detection of SARS-CoV-2 RNA that are detectable in nasal, nasopharyngeal, and oropharyngeal swab samples during infection.
Positive results are indicative of the presence of SARS-CoV-2 RNA; clinical correlation with patient history and other diagnostic information is necessary to determine patient infection status. Positive results do not rule out bacterial infection or co-infection with other viruses.
Laboratories within the United States and its territories are required to report all positive results to the appropriate public health authorities.
Negative results do not preclude SARS-CoV-2 infection and should not be used as the sole basis for patient management decisions. Negative results must be combined with clinical observations, patient history, and epidemiological information. (Source.)
The media has also been reporting a shortage of COVID-19 test kits. The CDC has made recommendations on prioritizing patients based on need.
The media makes it appear that anyone can get a nasal swab and get tested. All you need is the result from that swab and you can find out if you are positive or negative for COVID-19 as this Fox News Report implies:
This is far from the truth.
This article by ARUP Laboratories: “How Accurate Are COVID-19 Tests? Many Factors Can Affect Sensitivity, Specificity of Test Results” explains.
Due to internet censorship, articles on research that don’t conform to the current narrative are difficult to find but not impossible. The individual research papers are available by accessing the provided links.
The samples that were tested as part of these research papers were performed in a closed environment, meaning, collection, testing and results interpretations were all done on the same site.
What happens to the validity of test samples that are collected at one location, sent to a local health department, because of insufficient testing capabilities, sent to reference lab and from there sent to an affiliate lab location?
The CDC website has guidelines here.
In summary, the government and the media used fictional statistics on the severity of the COVID-19 “pandemic” to instill fear in the US population, shut down entire industries and trash a thriving economy.
States’ governors forced draconian “shelter in place” orders falsely claiming the necessity to “flatten the curve.”
These same governors are now claiming the need for mass, unproven antibody testing, those tests receiving FDA EUA, before states can “safely” open up again.
At least there is more reporting on the fallacies of antibody testing then there is on the testing for COVID itself.
Dr. Fauci is already warning of a COVID-19 resurgence this fall.
Source: Health Impact News
Subscribe for natural health news to your inbox. Follow Natural Blaze on YouTube, Twitter and Facebook. Become a Patron for as little as $1 per month.